医薬品開発の流れ
医薬品開発の流れ
医薬品開発は医薬品候補化合物を選定する基礎研究、動物を用いて医薬品候補化合物の有効性と安全性を確認する非臨床試験、ヒトにおける開発候補化合物の有効性と安全性を評価する臨床試験を実施した後に承認申請を行い規制当局の審査を経て承認に至ります。
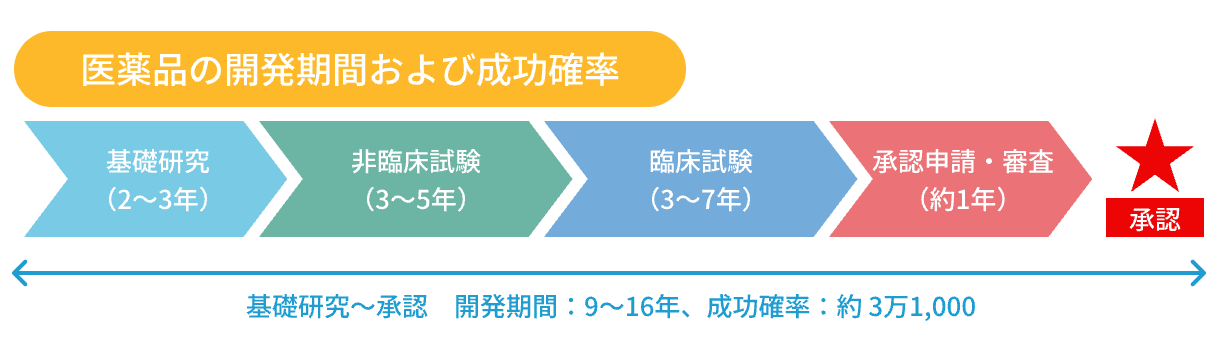
基礎研究段階は2~3年、非臨床試験段階は3~5年、臨床試験段階は3~7年、承認申請から承認まで約1年を要し、基礎研究から承認取得まで9~16年、成功確率は約 1/3万1,000と言われています。
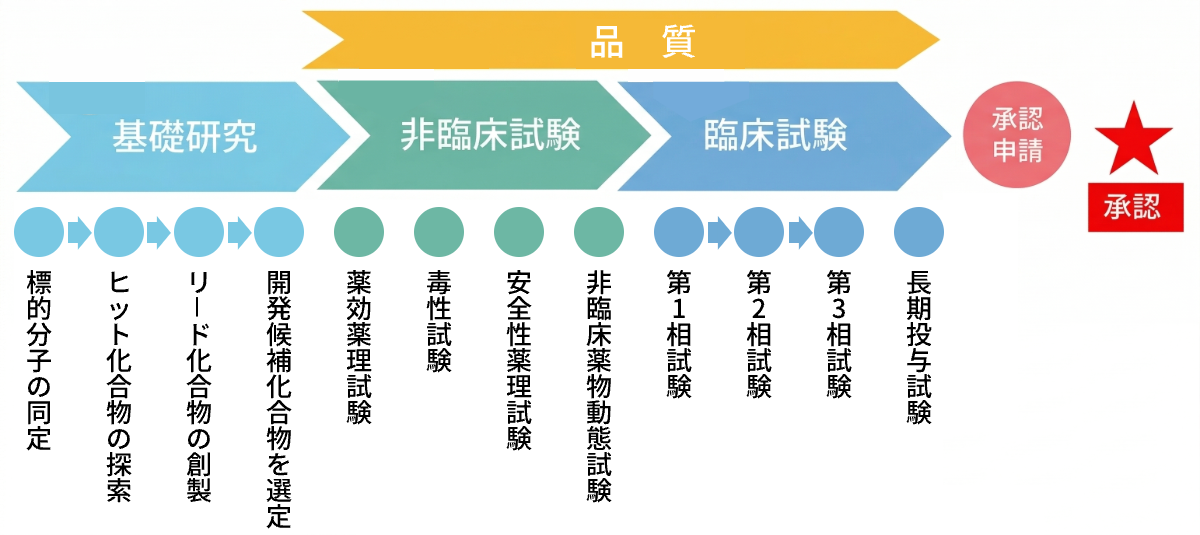
1.基礎研究
医薬品の候補になる開発候補化合物を選定します。
①標的分子の同定
医薬品開発のターゲットとする疾患のメカニズムを解明し標的分子を同定します。
②ヒット化合物の探索
標的分子が同定された後に標的分子に活性を示すヒット化合物を化合物スクリーニングにより探索します。
③リード化合物の創製
ヒット化合物の活性を合成、構造最適化等により活性向上を図ったリード化合物を創製します。
④開発候補化合物を選定
リード化合物を薬理活性、薬物動態、製造などの観点で最適化を図り開発候補化合物を選定します。
2.非臨床試験
ヒトに投与する前に動物等を対象に薬効薬理試験、安全性薬理試験、毒性試験、非臨床薬物動態試験を実施して、開発候補化合物の有効性および安全性を評価します。
2-1.薬効薬理試験
①効力を裏付ける試験
疾患のモデル動物を用いて開発候補化合物の作用機序を明らかにするとともに薬効を確認します。試験報告書は製造販売承認申請資料のCTD「4.2.1.1効力を裏付ける試験」に添付します。
②副次的薬理試験
開発候補化合物が目的とする薬理作用以外の意図しない薬理作用・作用機序を確認します。試験報告書は製造販売承認申請資料のCTD「4.2.1.2副次的薬理試験」に添付します。
2-2.安全性薬理試験
治療用量及びそれ以上の曝露に関連した被験物質の生理機能に対する潜在的な望ましくない薬力学的作用を検討します。
安全性薬理試験は「安全性薬理試験ガイドライン」に基づき、GLP(医薬品の安全性に関する非臨床試験の実施の基準に関する省令)を遵守して実施します。ヒト初回投与試験における被検者の安全性確保等のために初回の治験計画の届出の際には安全性薬理試験の最終報告書の提出が必要になります。安全性薬理試験の実施時期に関しては「医薬品の臨床試験及び製造販売承認申請のための非臨床安全性試験の実施についてのガイダンス」で規定されています。試験報告書は製造販売承認申請資料のCTD「4.2.1.3安全性薬理試験」に添付します。
2-3.毒性試験
開発候補化合物の安全性を単回投与毒性試験、反復投与毒性試験、癌原性試験、遺伝毒性試験、生殖発生毒性試験等を実施して評価します。
毒性試験は「医薬品毒性試験法ガイドライン」および「単回及び反復投与毒性試験ガイドラインの改正」に基づき、GLP(医薬品の安全性に関する非臨床試験の実施の基準に関する省令)を遵守して実施します。ヒト初回投与試験における被検者の安全性確保等のために初回の治験計画の届出の際には毒性試験の最終報告書の提出が必要になります。毒性試験の実施時期等に関しては「医薬品の臨床試験及び製造販売承認申請のための非臨床安全性試験の実施についてのガイダンス」で規定されています。
試験報告書は製造販売承認申請資料のCTD「4.2.3毒性試験」に添付します。
2-4.非臨床薬物動態試験
動物及びin vitro試験系を用いた非臨床薬物動態試験で開発候補化合物の体内動態 (吸収、分布、代謝、排泄)を明確にします。
非臨床薬物動態試験では最高血中濃度(Cmax)、最高血中濃度到達時間(Tmax)、血中濃度時間曲線下面積(AUC)、消失半減期(又はこれに準じた定数)、クリアランス、分布容積、生物学的利用性等のパラメータを求めるとともに、体内動態の非線形性の有無を検討します。また、必要に応じ、代謝物についても検討します。
薬物動態試験は「非臨床薬物動態試験ガイドライン」および「反復投与組織分布試験ガイダンス」に基づき実施します。
試験報告書は製造販売承認申請資料のCTD「4.2.2薬物動態試験」に添付します。
3.品質に関する試験
開発候補化合物の合成方法等を確立し、製剤検討を行い剤型の検討を行います。品質を確保するための規格及び試験方法を確立します。安定性試験を実施して治験薬/製品の有効期間を設定します。
原薬および製剤の品質確保はGMP(医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令)を遵守して実施します。
品質に関する試験の報告書は製造販売承認申請資料のCTD「Module 3品質に関する文書」に添付します。
4.臨床試験
ヒトを対象にして開発候補化合物の有効性および安全性を評価します。
4-1.第1相試験
少数の健康成人を対象に開発候補化合物の安全性および薬物動態(吸収、分布、代謝、排泄)について単回投与試験、反復投与試験により確認します。第2相試験の投与方法を決めるために食事の影響を第1相試験で確認することもあります。
抗悪性腫瘍剤の第1相試験は主にがん患者を対象に医薬品の候補化合物の安全性、忍容性および薬物動態を評価します。
4-2.第2相試験(探索的試験)
少数の患者を対象に開発候補化合物の有効性の探索と用法・用量の設定をおよび安全性を評価します。第2相試験では用量反応関係を確認するために第1相試験で安全性を確認した投与量の範囲内で複数の用量を用います。
4-3.第3相試験(検証的試験)
多数の患者を対象に第2相試験の結果に基づき設定した仮説を検証するとともに安全性を評価します。一般的にはプラセボを対照群として医薬品の候補化合物がプラセボに比べて有意に優れていることを示す優越性試験として実施します。なお、既に承認されている薬剤を対照群として医薬品の候補化合物が既承認薬剤に比べて劣っていないことを示す非劣性試験として実施することもあります。
4-4.長期投与試験
致命的でない疾患の治療のために長期間の投与(6カ月以上、継続的または繰り返し間歇的)が想定される場合は長期投与試験により安全性を評価します。
臨床試験はGCP(医薬品の安全性に関する非臨床試験の実施の基準に関する省令)を遵守して実施します。
臨床試験に関する報告書は製造販売承認申請資料のCTD「Module 5臨床試験報告書」に添付します。